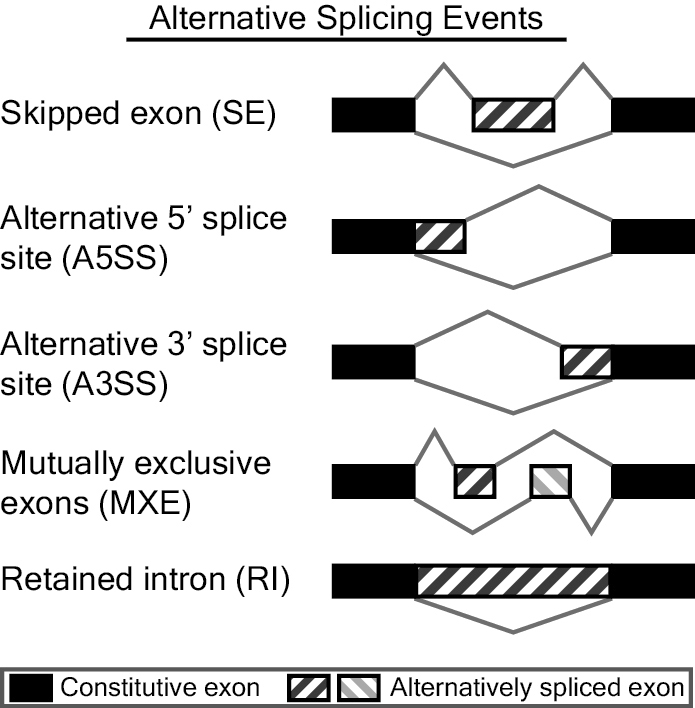
rMATS是一款对RNA-Seq数据进行差异可变剪切分析的软件。其通过rMATS统计模型对不同样本(有生物学重复的)进行可变剪切事件的表达定量,然后以likelihood-ratio test计算P value来表示两组样品在IncLevel(Inclusion Level)水平上的差异(从公式上来看,IncLevel跟PSI的定义也是类似的),lncLevel并利用Benjamini Hochberg算法对p value进行校正得FDR值。rMATS可识别的可变剪切事件有5种,分别是skipped exon (SE)外显子跳跃,alternative 5' splice site (A5SS)第一个外显子可变剪切,alternative 3' splice site (A3SS)最后一个外显子可变剪切,mutually exclusive exons (MXE)外显子选择性跳跃和 retained intron (RI)内含子滞留,展现形式如下图(来自官网http://rnaseq-mats.sourceforge.net/index.html)
 将reads比对至Reference上是采用STAR的STAR 2-pass模式,所以为了学习该教程,必须先学习如何使用STAR了
将reads比对至Reference上是采用STAR的STAR 2-pass模式,所以为了学习该教程,必须先学习如何使用STAR了