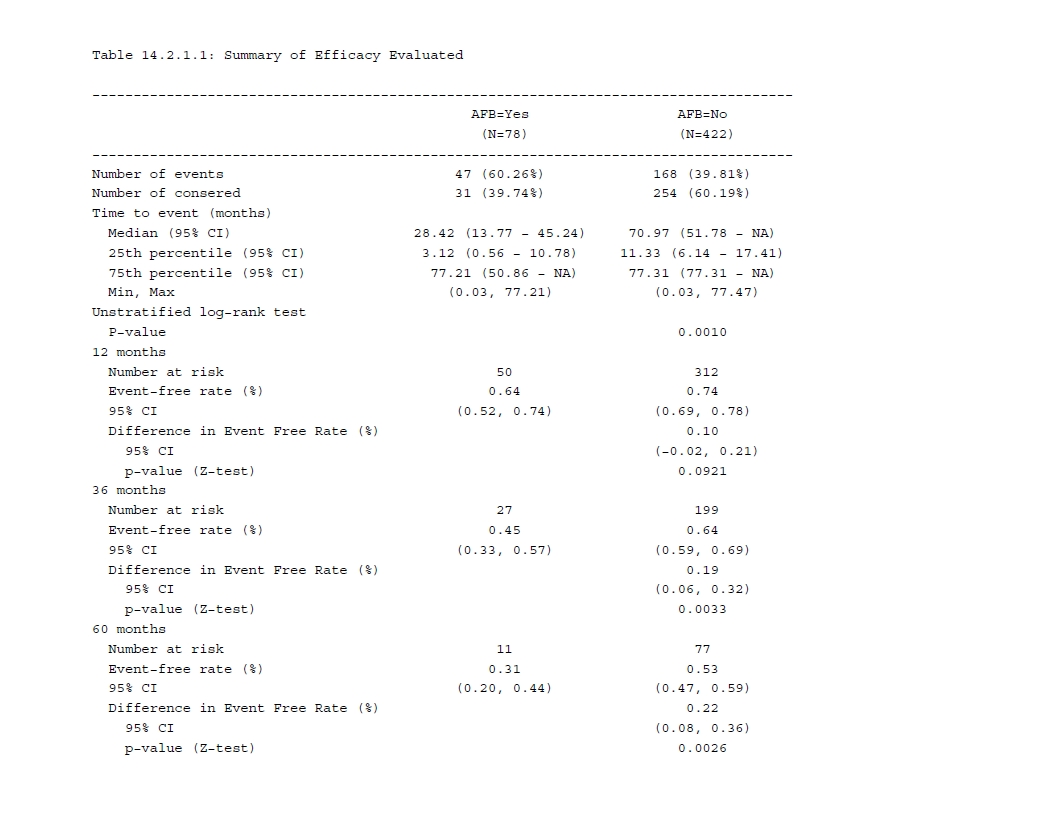
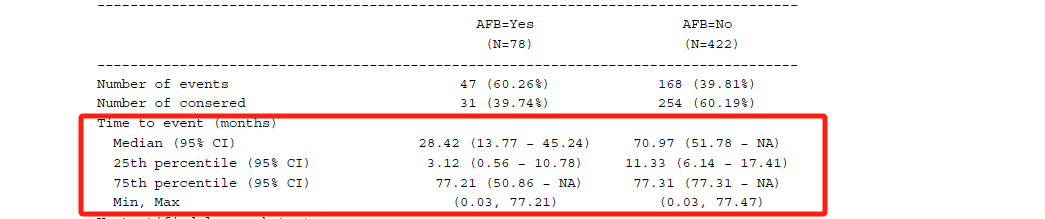
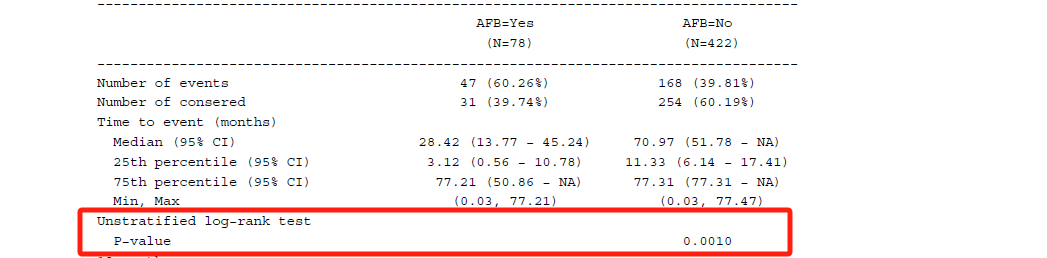
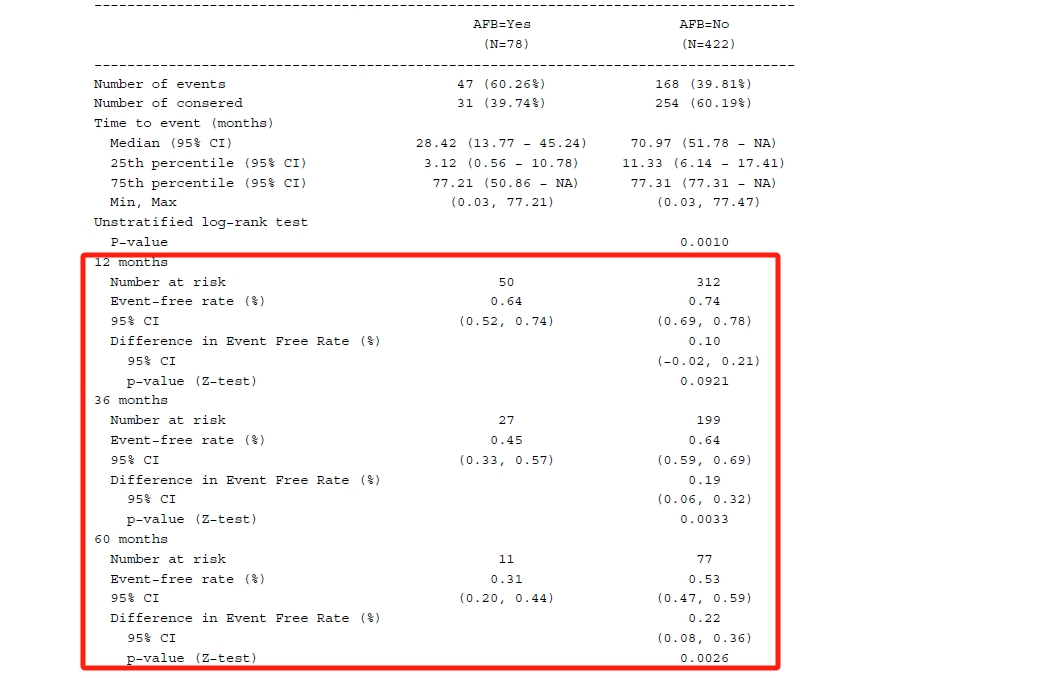
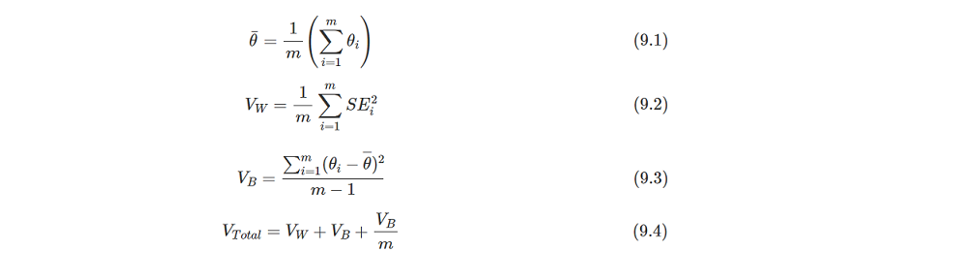In some ways, tipping point analysis can be regarded as supplementary to multiple imputation, allowing us to explore the outcomes from different scenarios by applying the shift parameters, such as decreasing the treatment effect while increasing the control group effect. The end goal is to identify the point at which the primary analysis result with multiple imputation becomes non-significant.
The tipping point analysis can be easily implemented using SAS procedure PROC MI and the brief instructions can be found in Sensitivity Analysis with the Tipping-Point Approach where it uses the MONOTONE statement to impute the missing data and the MNAR statement to adjust the imputed data by a range of specified shift parameters. Thus the analysis steps can be described as follows:
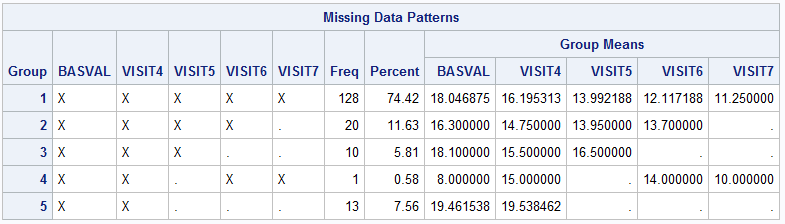
- If the missing pattern you detect is intermittent rather than monotone, you should fill in the missing data using MCMC statement so that the pattern can be transformed into monotone.
- If the missing data has monotone pattern, impute it using
MONOTONEorFCSmethod inProc MIunder the MAR assumptions. And specify the shift parameters inMNARstatement to adjust the imputed value for observations in any treatment group as needed. - Based on the imputed datasets from step 2, apply the pre-specified models to analyze each dataset and obtain the statistical results.
- Combine all of the results from step 3 by Rubin's rule using
Proc MIANALYZEand make statistical inferences. - Repeat steps 2-4 by adjusting the shift parameters to get a set of inference outcomes to identify the tipping point that overturns the conclusions from significant to non-significant.
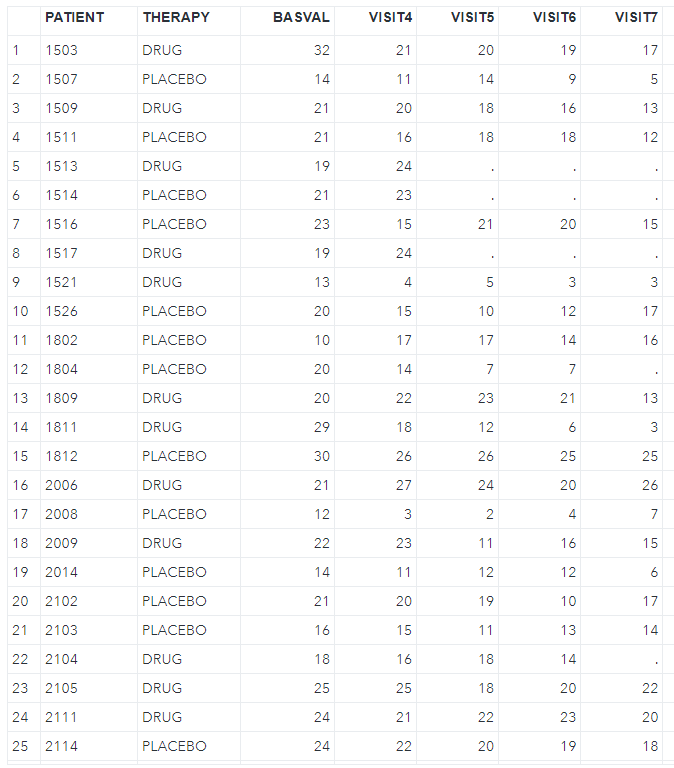
Here, let's look at the SAS code below. I will use the example missing dataset (low1.sas7bdat) from Mallinckrodt et al. (https://journals.sagepub.com/doi/pdf/10.1177/2168479013501310). And transform it to fit the follow-up analysis requirement.
proc sort data=low1; by patient trt basval; run;proc transpose data=low1 out=low1_wide(drop=_name_) prefix=week; by patient trt basval; id week; var change;run;As I have checked this dataset has a monotone missing pattern, so I don't need to go through step 1. But if you need or want to achieve that, try the code below. Please note that we have generated nimpute=10 imputed datasets in this step, we just need to apply nimpute=1 in the next monotone imputation and MNAR adjust steps.
/* Step 1: Achieve Monotone Missing Data Pattern */proc sort data=low1_wide; by trt; run;proc mi data=low1_wide seed=12306 nimpute=10 out=imp_mono; mcmc impute=monotone nbiter=1000 niter=1000; by trt; var basval week1 week2 week4 week6 week8;run;/* Step2: MAR imputation and MNAR adjustation at week 8 visit for each group. */proc sort data=imp_mono; by _imputation_ trt; run;proc mi data=imp_mono seed=12306 nimpute=10 out=imp_mnar2; class trt; by _imputation_ trt; var trt basval week1 week2 week4 week6 week8; monotone reg; mnar adjust (week8 / shift=-1 adjustobs=(trt='1')); mnar adjust (week8 / shift=1 adjustobs=(trt='2'));run;Back to assuming the example dataset has a monotone pattern, we just jump into step 2. Here, I suppose the primary endpoint is the change from baseline at week 8. So the prior visits are imputed under the MAR assumption using MONOTONE REG, and the week 8 visit will have an additional MNAR process where the trt=1 group is made better by adding a delta (shift=-1) while the trt=2 group is made worse by a delta (shift=1) as the lower value implies the better treatment effect. I also want to impute the datasets separately for each treatment group, so I set the BY statement to trt.
/* Step2: MAR imputation using MONOTONE */proc sort data=low1_wide; by trt; run;proc mi data=low1_wide seed=12306 nimpute=10 out=imp_mnar2; class trt; by trt; var basval week1 week2 week4 week6 week8; monotone reg; mnar adjust (week8 / shift=-1 adjustobs=(trt='1')); mnar adjust (week8 / shift=1 adjustobs=(trt='2'));run;From the SAS documentation, the MNAR statement is applicable only if it is used along with the MONOTONE and FCS statement. So why did I choose the former one here instead of the latter? Refer to this article (Application of Tipping Point Analysis in Clinical Trials using the Multiple Imputation Procedure in SAS), it states that only MONOTONE can provide us with the exact shift value we specified in imputed values straightforwardly, whereas the FCS needs a bit trick processing although it has more advantages somewhere. P.S. I did check it, indeed as mentioned above.
Now we generate 10 imputed datasets with a single shift value, and then these complete datasets are analyzed using the ANCOVA model, and the results are combined using Proc MIANALYZE, which is a typical multiple imputation process. So we don't need to describe them as details, just put all of above steps into one macro so that we can get a set of results using different shift values.
%macro mi_tpa(ind=, smin=, smax=, sinc=, out=); /* Create a set of shift values */ %let ncase=%sysevalf((&smax. - &smin.) / &sinc., ceil); data &out.; set _null_; run; /* Looping implement each shift for monotone imputation */ %do i=0 %to &ncase.; %let k=%sysevalf(&smin. + &i. * &sinc.); proc sort data=&ind.; by trt; run; proc mi data=&ind. seed=12306 nimpute=10 out=imp_mnar; class trt; by trt; var basval week1 week2 week4 week6 week8; monotone reg; mnar adjust (week8 / shift=-&k. adjustobs=(trt='1')); mnar adjust (week8 / shift=&k. adjustobs=(trt='2')); run; proc sort; by _imputation_; run; /* Step 3: Implement ANCOVA model for each imputation*/ ods output lsmeans=lsm diffs=diff; proc mixed data=imp_mnar; by _imputation_; class trt(ref='1'); model week8=basval trt /ddfm=kr; lsmeans trt / cl pdiff diff; run; /* Step 4: Pooling model results */ ods output ParameterEstimates=combined_diff; proc mianalyze data=diff; by trt _trt; modeleffects estimate; stderr stderr; run; /* Output results */ data mnar; set combined_diff; shift=&k.; run; data &out.; set &out. mnar; run; %end;%mend;Here, we assume that the tipping point that reverses the conclusion is between 0 and 5. Thus I define the range from 0 to 5 with an interval of 0.5. The following code performs the MNAR adjustment with each of the shift values, like 0, 0.5, 1,...,4.5, 5.
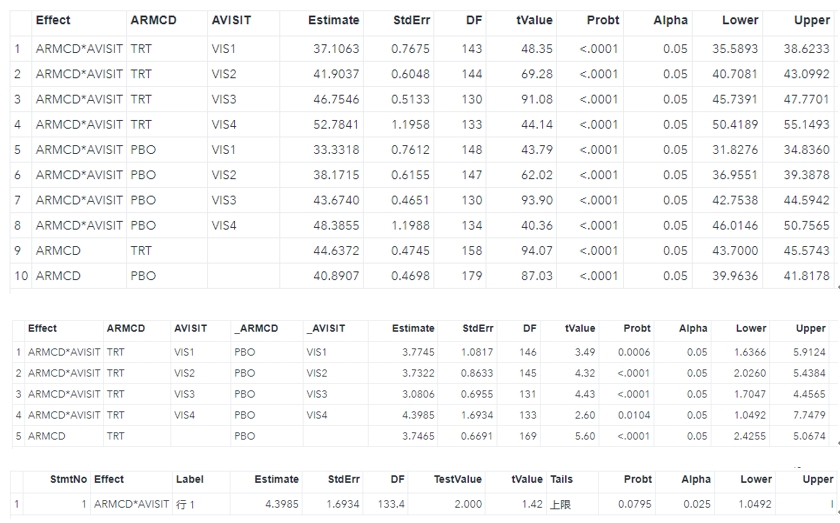
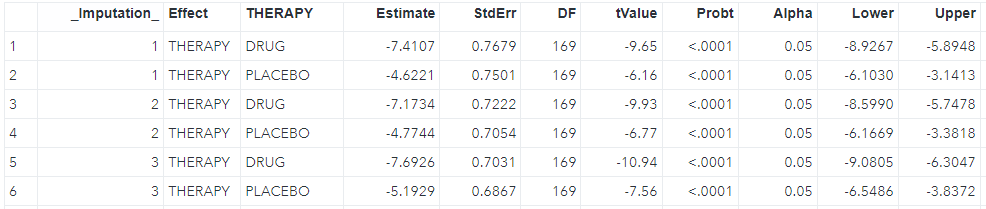
%mi_tpa(ind=low1_wide, smin=0, smax=5, sinc=0.5, out=tpa_rst);The output table can be shown below.

Regarding each shift value, the smallest value where the p-value is no longer significant is identified as the tipping point (in the red box). The next question is whether the shift value is reasonable in clinical practice. If that is not reasonable or unlikely, it can provide strong support for out primary conclusion.
Reference
TIPPING POINT ANALYSES IN MISSING DATA IMPUTATION
Application of Tipping Point Analysis in Clinical Trials using the Multiple Imputation Procedure in SAS
https://classic.clinicaltrials.gov/ProvidedDocs/91/NCT03282591/SAP_001.pdf
https://cdn.clinicaltrials.gov/large-docs/92/NCT03759392/SAP_001.pdf