Pathview是一个用于整合表达谱数据并用于可视化kegg通路的一个R包,其会先下载kegg官网上的通路图,然后整合输入数据对通路图进行再次渲染(加工?),从而对kegg通路图进行一定程度上的个性化处理 Pathview是一个bioconductor包,正常安装即可
source("https://bioconductor.org/biocLite.R")
biocLite("pathview")以其自带测试数据集,对Pathview包的可视化功能做个整理,用法很简单,主要为围绕着pathview这个作图函数(参数很多,用?pathview查看下各个参数的说明),只是需要将数据整理或者整合成其需要的输入数据格式即可
先看下demo数据是什么格式,列名是每个样本名,行名是每个gene的entrez id
> head(gse16873.d)
DCIS_1 DCIS_2 DCIS_3 DCIS_4 DCIS_5 DCIS_6
10000 -0.30764480 -0.14722769 -0.023784808 -0.07056193 -0.001323087 -0.15026813
10001 0.41586805 -0.33477259 -0.513136907 -0.16653712 0.111122223 0.13400734
10002 0.19854925 0.03789588 0.341865341 -0.08527420 0.767559264 0.15828609
10003 -0.23155297 -0.09659311 -0.104727283 -0.04801404 -0.208056443 0.03344448
100048912 -0.04490724 -0.05203146 0.036390376 0.04807823 0.027205816 0.05444739
10004 -0.08756237 -0.05027725 0.001821133 0.03023835 0.008034394 -0.06860749先看下Pathview最常见的一种用法:将某个样本的表达量(测试数据已经是归一化后的表达量);其实也可以任何列数据,不仅仅是表达量数据,也可以是foldchange等数据,人为特定的数值型数据也行;最后以color bar的形式在kegg通路图上的对应节点展示;如下例子所示,取第一个样本的数值最为gene.data,通路选择04110,物种为hsa
p <- pathview(gene.data = gse16873.d[, 1], pathway.id = "04110", species = "hsa", out.suffix = "gse16873")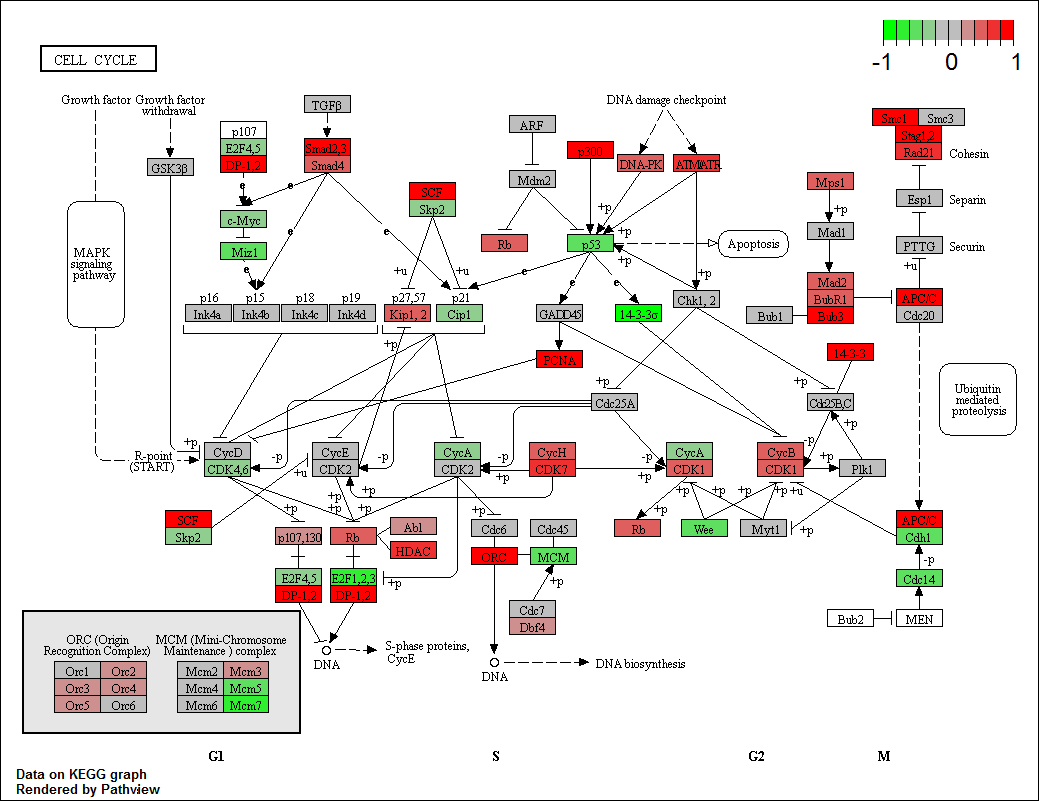
图中右上角color bar是可以通过参数limit来调整的,默认是limit=list(gene=1, cpd=1),即gene.data的限定范围在(-1,1),cpd.data的限定范围在c(-1,1);如果有其他需要可以设置为limit=list(gene=2, cpd=1)等;如果想最大值和最小值不对称则limit = list(gene=c(-1,2))即可
如果想将color bar分割的更加密集一些,则可以修改默认参数bins = list(gene = 10, cpd = 10)为20。。。只能说Pathview作者好细心。。
接下来看看Pathview如何展示整合数据的,如基因和代谢组的数据在kegg通路上整合可视化
先载入代谢物相关数据,如
sim.cpd.data=sim.mol.data(mol.type="cpd", nmol=3000)
> head(sim.cpd.data)
C02787 C08521 C01043 C11496 C07111 C00031
-1.15259948 0.46416071 0.72893247 0.41061745 -1.46114720 -0.01890809 然后通过cpd.data参数将代谢物数据加入到pathview函数中,如:
p <- pathview(gene.data = gse16873.d[, 1], cpd.data = sim.cpd.data,
pathway.id = "00640", species = "hsa", out.suffix = "gse16873.cpd",
keys.align = "y", kegg.native = T, key.pos = "topright")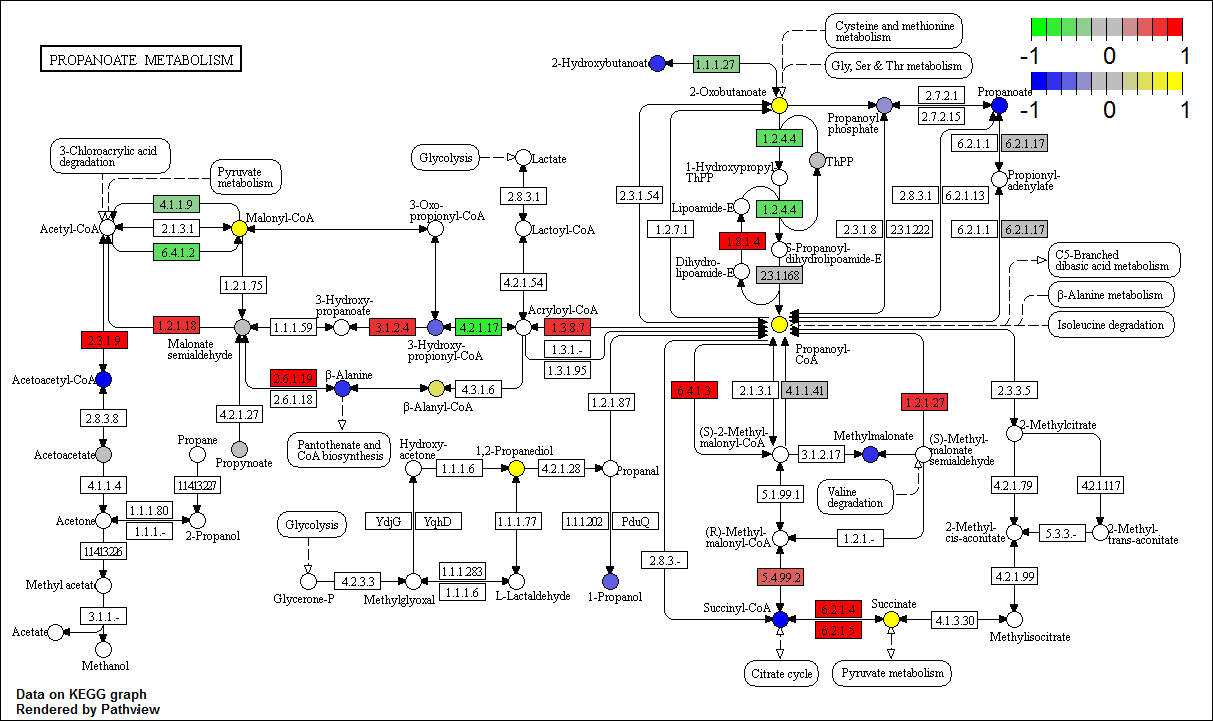
keys.align = "y"控制color bar对齐方式,key.pos = "topright"则是对应color bar在图上的位置(主要有bottomleft,bottomright,topleft以及topright)
Pathview不仅能整合不同组学数据(上述例子是转录组和代谢组),还可以整合多个样本的数据,比如我们在上面看到gene.data只导入了一列数据,其实Pathview支持多列(也就是多个样本)数据的导入,cpd.data也一样
如按照说明文档中的模拟一组多样本的代谢物表达谱数据(也似乎是归一化后的)
sim.cpd.data2 = matrix(sample(sim.cpd.data, 18000, replace = T), ncol = 6)
rownames(sim.cpd.data2) = names(sim.cpd.data)
colnames(sim.cpd.data2) = paste("exp", 1:6, sep = "")
> head(sim.cpd.data2, 3)
exp1 exp2 exp3 exp4 exp5 exp6
C02787 -0.5425224 1.7940544 -0.2629972 0.2729004 -0.4897083 1.05131740
C08521 -1.1903358 0.4448658 2.6074747 -0.9163451 0.1239377 0.57827710
C01043 0.3391817 1.6855815 1.0203767 -1.3184792 0.4727454 0.03381888然后还是使用pathview函数作图,相比上面例子增加了参数match.data = F,主要用于表示gene和cpd样本数是否要匹配;multi.state = T则表示多个样本在同一个图上显示
p <- pathview(gene.data = gse16873.d[, 1:3], cpd.data = sim.cpd.data2[, 1:2],
pathway.id = "00640", species = "hsa", out.suffix = "gse16873.cpd.3-2s",
keys.align = "y", kegg.native = T, match.data = F, multi.state = T, same.layer = T)
正如之前说的,Pathview不仅能展示连续性数据,还可以展示离散型数据(如:差异显著OR不显著,上调OR下调等),我先模拟一个(0,1)离散型数据
testdata2 <- data.frame(sample1=sample(c(0,1), 10000, replace = T), stringsAsFactors = F)
row.names(testdata2) <- row.names(gse16873.d)[1:10000]然后在pathview函数中将limit = list(gene = c(0,1))和bins = list(gene = 1)设置为一致;由于默认颜色是低(绿色),中(灰色),高(红色),而我们测试数据是0,1两个数字,所以需要将mid设置为红色mid = list(gene = "red"),这样0就是绿色,1就是红色了
p <- pathview(gene.data = testdata, pathway.id = "00640", species = "hsa",
out.suffix = "test.genes",discrete = list(gene = T), kegg.native = T,
limit = list(gene = c(0,1)), bins = list(gene = 1), mid = list(gene = "red"))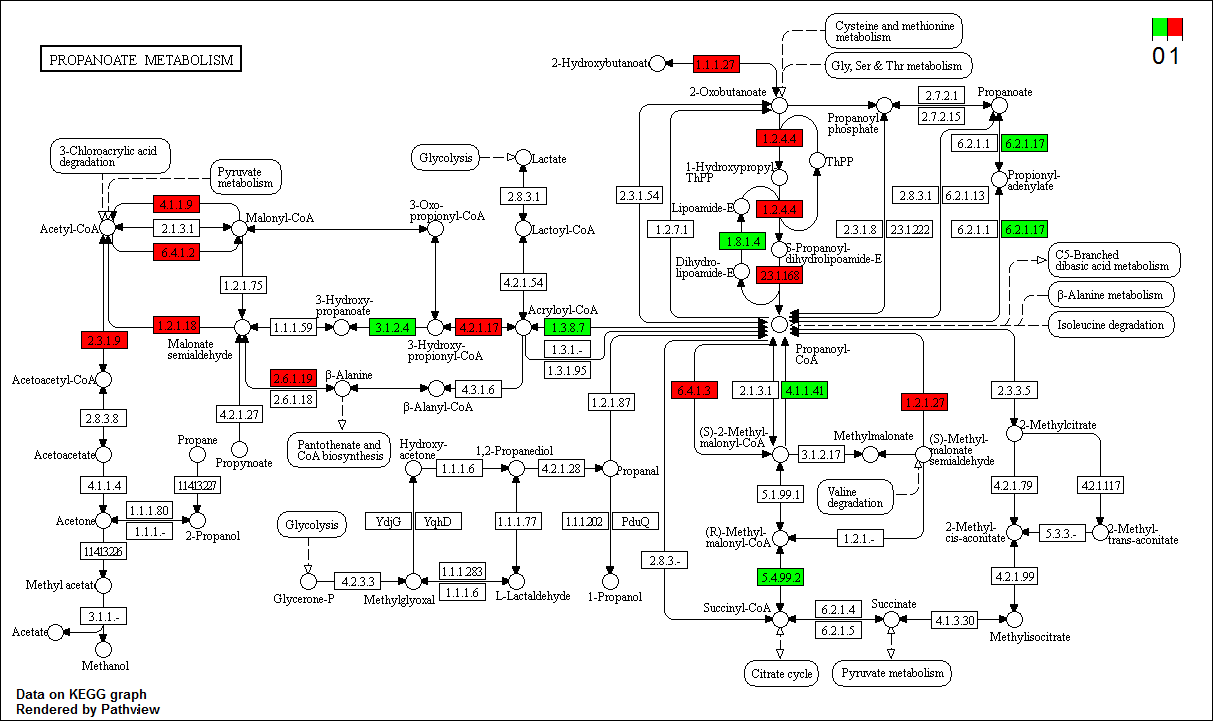
从我们之前的测试数据中可看到,pathview函数默认使用的Id是entrez id,从默认参数gene.idtype="entrez"可得;对于绝大多数真核生物来说,其entrez id确实等于其对应的kegg id,但是有些物种还是特例的,比如拟南芥。。。当然Pathview包给出了一些id转化方法(但我一般不用,因为我比较喜欢自己来转化);因此一般我们能拿到的就已经是kegg id了,比如拟南芥的ath:AT4G17260,这是需要设置gene.idtype=kegg,这里需要注意的,gene.data参数输入的不是ath:AT4G17260而是AT4G17260,下面以一个例子说明
先模拟一个数据
testid <- data.frame(expr = rnorm(5), stringsAsFactors = F)
row.names(testid) <- c("AT4G17260", "AT2G14170", "AT2G20420", "AT1G06550", "AT1G36160")然后还是以通路00640为例,物种是拟南芥
p <- pathview(gene.data = testid, pathway.id = "00640", species = "ath",
gene.idtype = "kegg", out.suffix = "testid")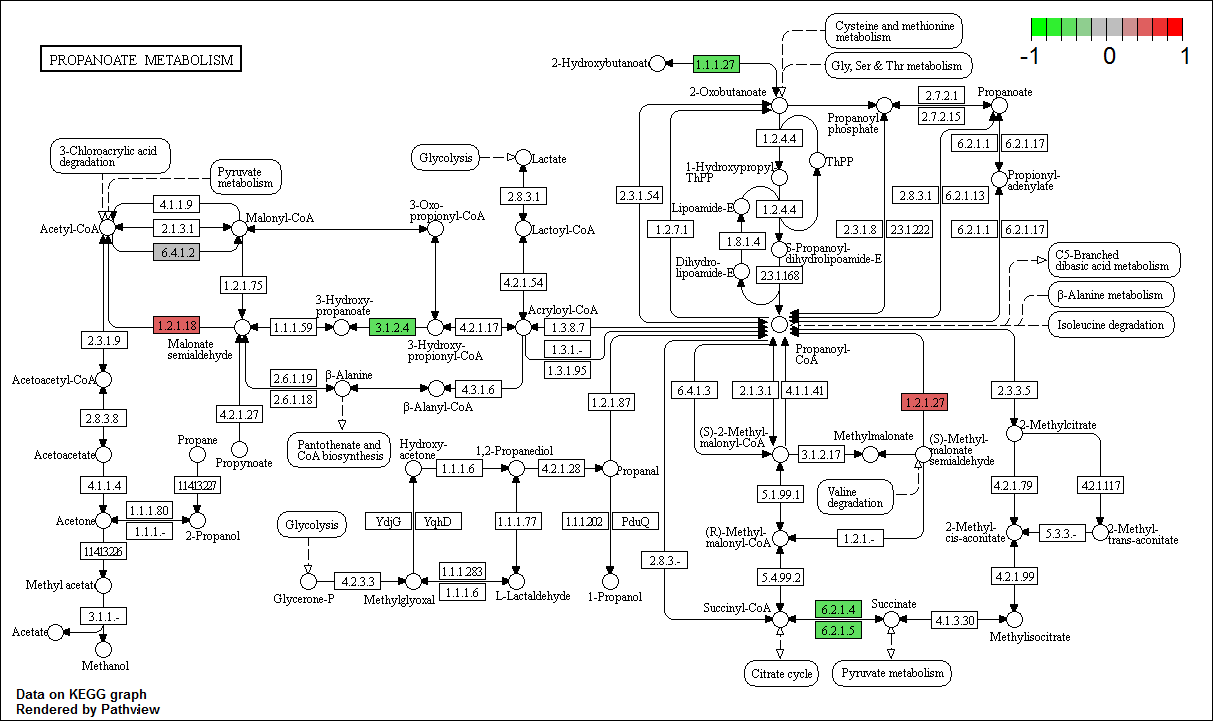
但是当物种是kegg未收录的物种,这是可能用的不再是kegg id还是K号,如K00016,这时的通路则是KEGG ortholog pathways,物种设定为ko,即:species = "ko"
testko <- data.frame(expr = rnorm(5), stringsAsFactors = F)
row.names(testko) <- c("K00016", "K01026", "K00140", "K18371", "K17741")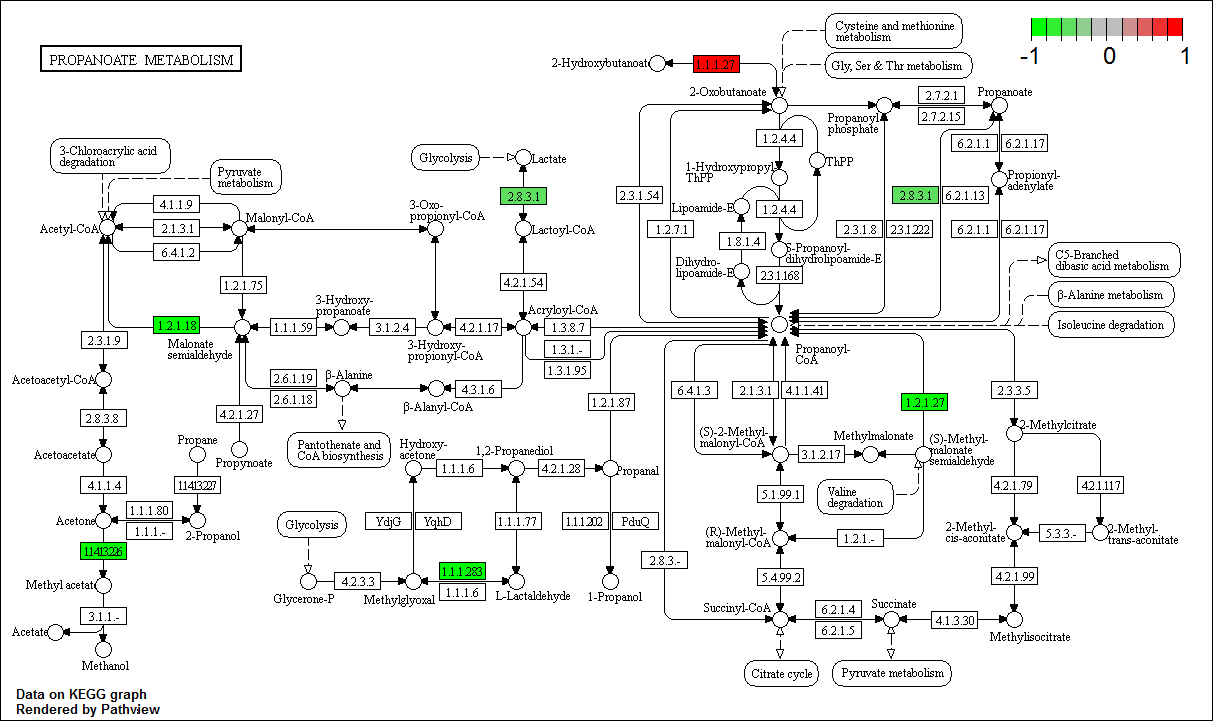
上述大概是Pathview包主要的用法了,如果有需要可以再从头看下其说明文档http://bioconductor.org/packages/release/bioc/vignettes/pathview/inst/doc/pathview.pdf
本文出自于http://www.bioinfo-scrounger.com转载请注明出处