最近看到一篇文献Sex-specific transcriptional signatures in human depression,其中提到了用Rank-rank hypergeometric overlap (RRHO)方法来寻找不同类型样本之间的overlap基因
文章研究在Major depressive disorder(MMD)下,对6个brain regions(vmPFC, OFC, dlPFC, aINS, NAc, vSUB)进行转录组测序,研究不同性别下与MMD相关的6个brain regions的一些表达特征;通过RRHO来寻找brain regions的overlapping patterns,相当于找一些有共同影响作用的基因 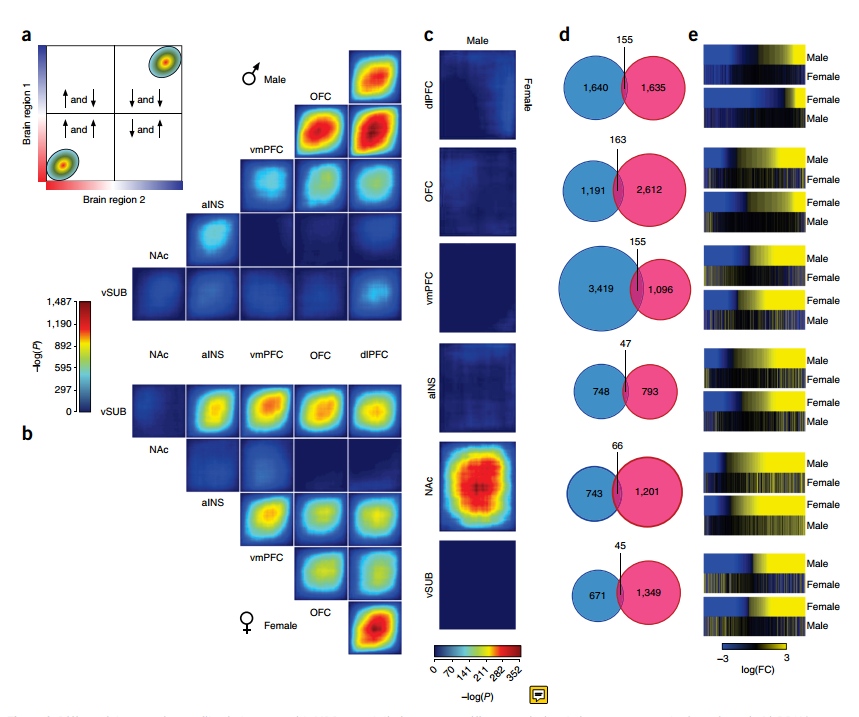
文章不做解读,但是这个RRHO方法的思路觉得有点意思,其出自文献Rank–rank hypergeometric overlap: identification of statistically significant overlap between gene-expression signatures
RRHO方法是将不同class样本的基因表达谱进行排序(两个class),然后利用超几何分布迭代计算所有组合的P值,进而找到最佳overlapping基因组合;这方法不需要预先人为设定阈值(比如差异基因阈值),所以是unbiased
我是通过其R包-RRHO的源码理解其计算思路,然后结合上述方法学文章加以理解;其R包源码很简单(500行不到的代码),大致有以下几个步骤:
- 对每组class样本计算其gene ranking;这个类似于GSEA中的ranking,可选的方法也一样,下图中用的是tTest,也可以用signal-to-noise等等;然后可以根据ranking绘制scatter plot,如果两个class之间一致性很高,那会有一条很好的线性回归线(对角线)
- 对上述ranking按照降序排列,设置一个step(相当于基因组合的长度),穷尽两个class的组合,用的是
expand.grid函数 - 循环每个组合,计算每个组合的overlapping gene数以及通过超几何分布(
phyper函数)计算每个组合的P值;下图的H(k,s,M,N)中k代表overlapping gene数,s和M分别代表两个class中每个组合的gene数,N代表总gene数 - 最后则是对于P值进行校正,默认是用BY(Benjamini-Yekutieli)方法进行多重检验校正;此外还有用Permutation(置换检验)方法(如果每组class的样本足够的多,至少6个样本)来评估hypergeometric map的整体统计学意义,这里跟GSEA也比较相似,既可以选择sample permutation也可以选择gene permutation,但是文章作者建议使用前者(PS. R包则默认使用后者。。。)
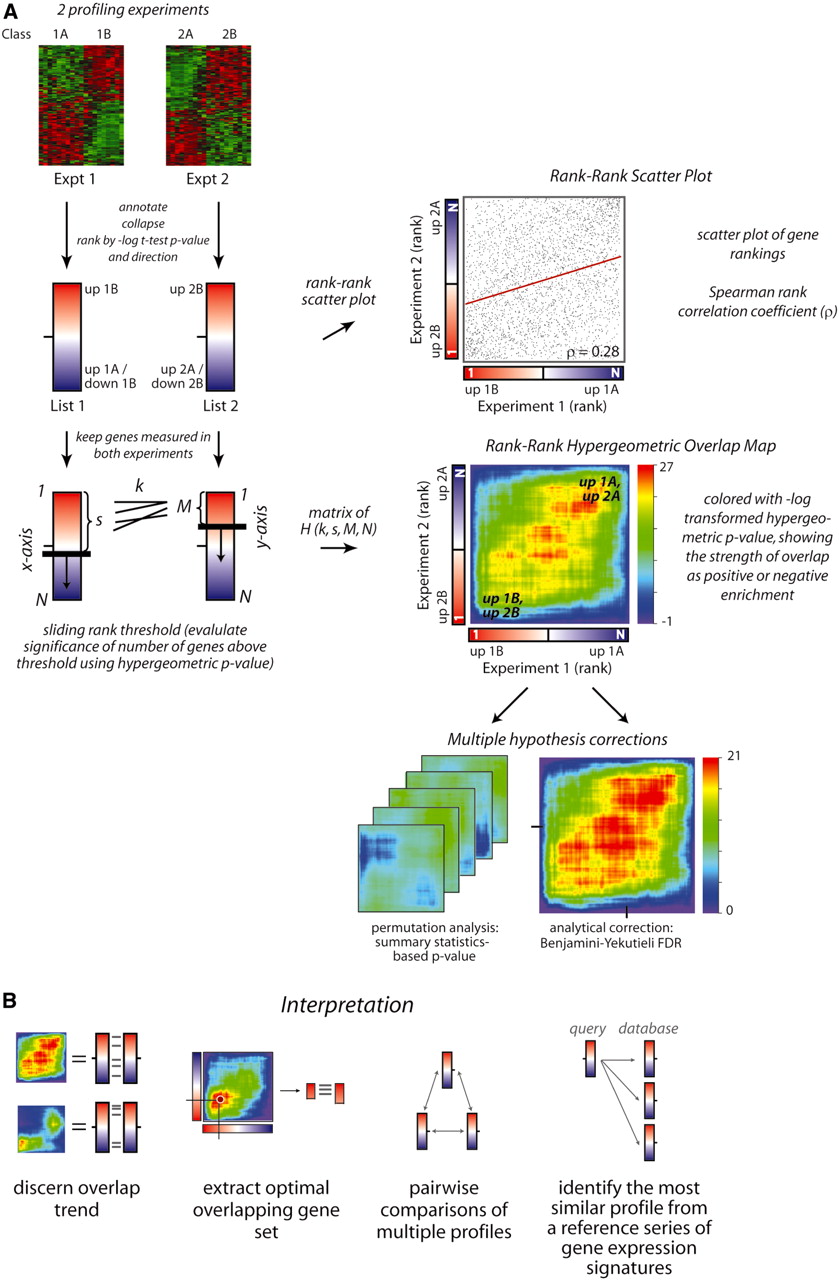
RRHO的结果怎么看呢,文章也给出了以下几点建议:
- Different map patterns indicate different types of overlap, such as the full profiles being correlated or only genes increasing in both experiments overlapping(看看整体表达趋势或者表达量同增的模式)
- The highest intensity point on the map can be used to extract the most statistically significant overlapping gene set(如果看到RRHO maps上有异常明显overlaping点,可以提取该set作为显著overlapping基因)
- To compare relative overlap pairwise within a set of profiles(就像最开始那篇文章用的,将多个brain regions的RRHO maps整合在一起比较)
- To compare an experimental profile to a series of reference signatures(将RRHO结果与其它reference signatures做比较)
其实已有人对RRHO方法做了整理,如一篇公众号文章超几何分布,RRHO--数据驱动寻找重叠基因,里面以PPT形式展示
生物信息分析方法繁多,各式各样的都有,所以有些方法不一定要用它,但是理解一下其思路也是不错的选择
本文出自于http://www.bioinfo-scrounger.com转载请注明出处