由于开始时文献阅读的不多,导致对一些甲基化芯片文章的理解造成了一定的偏差,这篇笔记陆陆续续改了几次
之前甲基化入门学习时本打算重复下提纲给的文献,但是后来学习过程中发现GEO上下载的RAW文件里没有该样本信息文件,就用了ChAMP包的测试数据。最后想了想,还是决定找一篇比较简单的文献的来实践使用下甲基化450K芯片的分析过程。看了几篇关于人的甲基化文献(数据在GEO上的),挑了一篇Intrinsic gene changes determine the successful establishment of stable renal cancer cell lines from tumour tissue,样本数据比较少,但是文章对分组比较明确(或者说是分的比较简单),所以拿来试试 GEO数据下载地址https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE89648
文章主要讲的内容理的不是太清楚,大概是作者报道了新的7个ccRCC稳定细胞系,可以稳定培养20代以上,并与finite细胞系(培养不超过10代的)做比较,进行表达芯片分析和甲基化芯片分析,从而找出一些关键基因在两组样本中差异表达,而这表达水平的变化可能也与甲基化水平有关,以其中一个基因SLC34A2做了重点研究及说明。
从文中我们可以看到作者对12个样本进行了分组,但是在基因表达芯片分析时是每组5个样本,P组为UT14(P2), UT16(P2), UT33a(P3),UT48(P2) and UT56(P1),N组为UT13a(P3),UT27(P3), UT37(P2), UT41(P2) and UT54(P3);但是由于UT16 and UT27两个样本的数据出现比较偏差,所以在两组中剔除了,所以最终分组是P组和N组各4个样本。
最后根据差异分析,P组相比N组,以fold change >2, p<0.05标准筛选出52个上调基因和65个下调基因,从中选出12个差异最为明显的基因去做了RT-PCR验证,最后也做了常规的GO富集分析。表达谱数据分析这里就不重复了,是Affymetrix的Affymetrix Human Transcriptome Array 2.0表达谱芯片,可用oligo包进行分析,可参照之前的博文回顾基因表达芯片分析(数据分析)
甲基化芯片的分组也跟表达芯片一样,两组各4个样本,在GEO网站上找到其对应的GSM号,然后参照ChAMP包给的测试例子中的lung_test_set.csv作为样式,手动做个sample.csv,以便能让ChAMP包识别,如下:
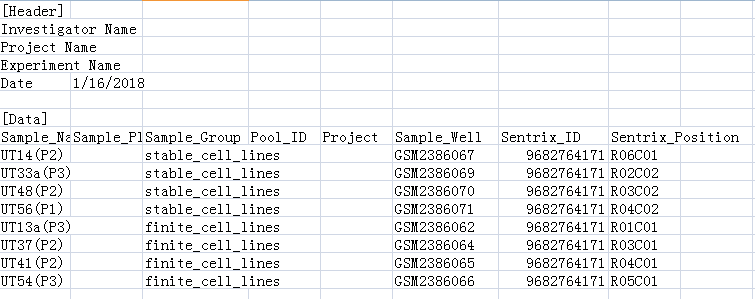
接下来就是用ChAMP包的champ.load()函数读入数据(数据放在450K目录下),做个函数不仅做了数据的读入,还做了数据的过滤,由于作者是用GenomeStudio v1.9 software(illumina的官方软件)来做处理的,过滤标准没写,而|β-Difference|>0.2 and p values <0.05则是作者定义差异甲基化位点的筛选标准,所以我就使用champ.load()的默认参数来过滤了
myLoad <- champ.load("./450K/",arraytype="450K")接着做下质控,看看有无异常样本啥的,还有densityPlot,每个样本的beta值的分布图,样本的聚类图等;一般是没什么大问题的
champ.QC(beta = myLoad$beta, pheno = myLoad$pd$Sample_Name)
QC.GUI(beta=myLoad$beta,arraytype="450K")接着是标准化,文章并没有说用什么标准化方法,我看的其他文章有些会提,比如DNA Methylation signatures in panic disorder这篇文章中就提到用的是BMIQ方法进行标准化;PS.其实这篇文章更加适合做练习,其在GEO上也有可下载的IDAT数据,作者是用minfi包做的分析,也列出了探针过滤的指标,分析过程也更加完善;但是作者是根据临床数据(比如性别、年龄等)进行分析的,可惜我没找对其形状与样本的对应关系,所以也没办法重复了
myNorm <- champ.norm(beta=myLoad$beta,arraytype="450K",cores=5)
#默认是采用BMIQ由于这个测试例子的样本较少,一把是不会需要批次校正的,所以这里SVD plot检查下一般是没问题的,但如上面那篇文章,有上百个样本,所以容易有批次效应,所以那篇作者用Empirical Bayes’ method ComBat做了批次校正
champ.SVD(beta=myNorm,pd=myLoad$pd)做完上述步骤,接下来就是做甲基化探针差异分析,本文的作者也做了这步,筛选标准并没有列出,我试了下用adj.pvalue < 0.05卡的话,就没有差异的探针的了。。所以我还是先用pvalue < 0.01作为阈值先筛下差异的甲基化位点
myDMP <- champ.DMP(beta = myNorm,pheno=myLoad$pd$Sample_Group,adjPVal = 1)
myDMP_df <- filter(myDMP$stable_cell_lines_to_finite_cell_lines, P.Value < 0.05 & abs(deltaBeta) > 0.2)作者分别看了下P组中高甲基化和低甲基化的基因数目 >showed that there were 527 highly methylated genes and 511 genes with low methylation in cells of the P group
我粗略查了下,一般可以将bata值大于0.6的位点认为是fully methylated,而bata值小于0.2的位点认为是fully unmethylated;但是可能这篇文献不是这个意思,所以我就在筛完差异的结果中以两个组的差值大于0.2 and P组的beta值大于0.6 作为高甲基化筛选标准, or 小于-0.2当做高/低甲基化的筛选方法(这里当然是P组相对N组来说的)
myDMP_highly_pgroup <- filter(myDMP_df, deltaBeta < -0.2 & stable_cell_lines_AVG > 0.6)
length(unique(myDMP_highly_pgroup$gene))
#[1] 970
myDMP_lowly_pgroup <- filter(myDMP_df, deltaBeta > 0.2 & stable_cell_lines_AVG < 0.2)
length(unique(myDMP_lowly_pgroup$gene))
#[1] 864结果肯定显而易见的跟文献不一样。。。相差的有点多了,可能高/低甲基化的定义不太一样吧(我只是初学,理解的可能不到位)
然后作者重点选了CD44, SEPP1, SULT1C4, AKAP12 and SLC34A2等5个基因作为测定ccRCC cell lines的甲基化状态的指标;我也看了下我的结果,有3个基因(SLC34A2,CD44,SULT1C4)是在我的差异甲基化位点列表里的
作者分析了基因表达芯片和甲基化芯片得出了以上的结果,整体上属于简单的分析;有些文献还做了 differentially methylated region (DMR) analysis,使用ChAMP包的champ.DMR函数,顺便用DMR.GUI看看在哪些基因上
myDMR <- champ.DMR(beta=myNorm,pheno=myLoad$pd$Sample_Group,method="Bumphunter")
DMR.GUI(DMR=myDMR)Summary
我这个文献测试是属于入门级别的,甲基化芯片的应用不局限于此。而且我挑的是有IDAT的GEO数据,有些公共数据则是TXT形式的,还没碰过。除了GEO上的公共数据外,其实TCGA中的甲基化数据也是用芯片测的,值得我之后再去研究研究。对于甲基化芯片的处理现在还只会简单的使用ChAMP包,还有一些参数以及细节需要掌握
本文出自于http://www.bioinfo-scrounger.com转载请注明出处