在转录组定量分析时,如果采用的是alignment-based转录组定量策略,那么一般会使用的是HISAT2、STAR或者TopHat等比对软件。
接着则是对转录组进行定量,如果是基于基因水平的定量,我之前一般是采用HTSeq-count工具来获取每个基因上的count数。所谓count数,个人简单的理解为根据不同比对情况,将reads分配到各个基因上。HTSeq-coun对于多重比对的reads则是采取舍弃策略。当HTSeq-count选择默认参数(-m 默认模式),那么reads是以下图所示的union的情况进行分配的 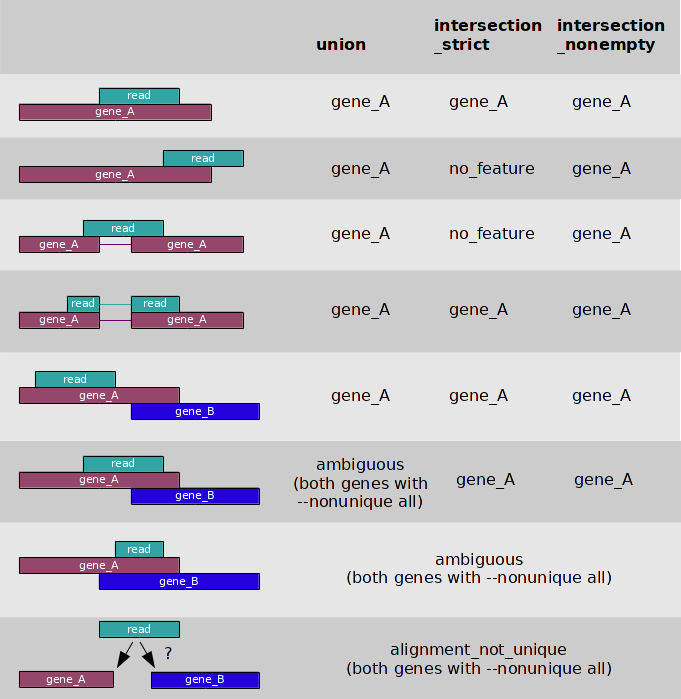
除了HTSeq-count工具外,其实也可以使用bedtools工具的multicov进行简单的基因水平定量。其需要一个所有基因的位置信息的bed文件,然后计算比对结果bam文件中的reads出现在基因interval上的个数,功能比较简单(说白了就是基于reads位置信息),适用性和准确性一般来说是没有HTSeq-count好。
以上两款在基因水平定量的软件有一个共同点,就是慢!如果样本数上升到几十及上百个样时,其所消耗的时间是以天记的,这时则可以考虑用featureCounts这款软件了,缺点还不知道,但是其优点已经听到过N遍了,就是快。。。
软件下载及安装
下载肯定去官网下载http://subread.sourceforge.net/
featureCounts: a ultrafast and accurate read summarization program
现在featureCounts已经整合在Subread里面了,粗略看了下简介和pdf文档,发现Subread是个功能很全面的软件,而且还有相对应的R包Rsubread,有机会再去瞧瞧。Subread有二进制版本,那么直接下载解压即可使用了
软件的使用
软件的作者认为其软件的优点在于(我就复制黏贴了):
- It carries out precise and accurate read assignments by taking care of indels, junctions and structural variants in the reads
- It takes only half a minute to summarize 20 million reads(真是快。。。)
- It supports GTF and SAF format annotation
- It supports strand-specific read counting
- It can count reads at feature (eg. exon) or meta-feature (eg. gene) level(也可以基于外显子定量)
- Highly flexible in counting multi-mapping and multi-overlapping reads. Such reads can be excluded, fully counted or fractionally counted(这点跟HTSeq-count不一样了,其对于多重比对的reads并不是采用全部丢弃的策略,按照其说法是更加灵活的对待)
- It gives users full control on the summarization of paired-end reads, including allowing them to check if both ends are mapped and/or if the fragment length falls within the specified range(可让使用者更加个性化的使用)
- Reduce ambuiguity in assigning read pairs by searching features that overlap with both reads from the pair
- It allows users to specify whether chimeric fragments should be counted(考虑的有点周到)
- Automatically detect input format (SAM or BAM)
- Automatically sort paired-end reads. Users can provide either location-sorted or namesorted bams files to featureCounts. Read sorting is implemented on the fly and it only incurs minimal time cost
featureCounts的参数有点多,可用-h先查看下
~/biosoft/subread/subread-1.6.0-Linux-x86_64/bin/featureCounts -h按照官网教程简单使用下:
~/biosoft/subread/subread-1.6.0-Linux-x86_64/bin/featureCounts -T 6 -p -t exon -g gene_id -a ~/annotation/mm10/gencode.vM13.annotation.gtf -o SRR3589959_featureCounts222.txt SRR3589959.bam主要的参数:
-a 输入GTF/GFF基因组注释文件
-p 这个参数是针对paired-end数据
-F 指定-a注释文件的格式,默认是GTF
-g 从注释文件中提取Meta-features信息用于read count,默认是gene_id
-t 跟-g一样的意思,其是默认将exon作为一个feature
-o 输出文件
-T 多线程数
其他参数介绍只能看文档了,不常用的话也是记不住的,要用时再去翻就行
运行中和运行后有两张图可以看看,主要讲了其运行中的一些信息,如下:
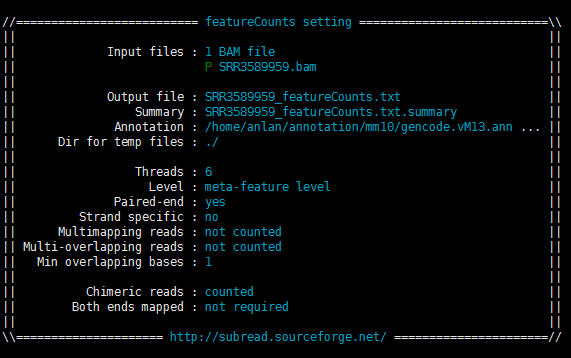
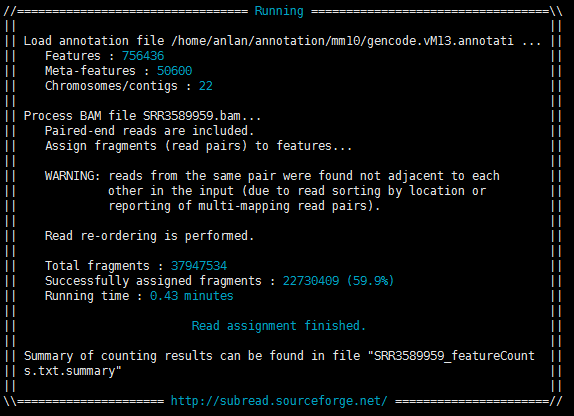
从Multimapping reads和Multi-overlapping reads均为not counted可看出我的运行参数是不考虑multimapping的情况的,并且如果一个read在多个基因上有overlaps(multi-overlapping),则软件也是不计入count数的(只对RNA-SEQ数据,ChIP-seq数据则是例外)
Min overlapping bases为1则说明--minOverlap参数默认是1,因而只考虑全部overlap的reads
Successfully assigned fragments : 22730409 (59.9%)则说明只有59.9%的paired reads定量到了基因上,如果想具体看看那些没有分配上的reads是出于原因,则可看输出中的summary文件
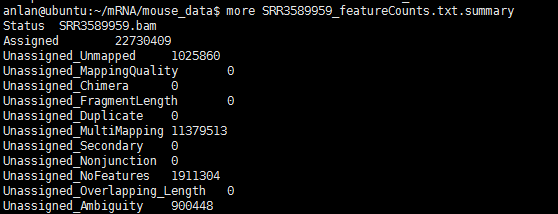
上图的相关说明可以看软件的pdf文档,其实也蛮好理解的
Running time : 0.33 minutes只能说真是快。。。
SRR3589959_featureCounts222.txt文件包含了featureCount结果的信息,如果想了解每个基因上的count数,则只需要提取出第1列和第7列的信息
cut -f 1,7 SRR3589959_featureCounts.txt |grep -v '^#' >feacturCounts.txt然后统计下featureCounts结果总count数和HTSeq-count结果的总count数(这个之前转录组学习时已经跑过了)
perl -alne '$sum += $F[1]; END {print $sum}' feacturCounts.txt
#22730409
grep -v '^_' HTSeq-count.txt |perl -alne '$sum += $F[1]; END {print $sum}'
#21514247从结果上看,两者数量相差还是蛮小的,看样子两者软件的默认参数的对待reads的方式还是差不多的(主要featureCounts的默认参数对待Multimapping reads是相同的,因为从图3可看出,MultiMapping的reads还是蛮多的)
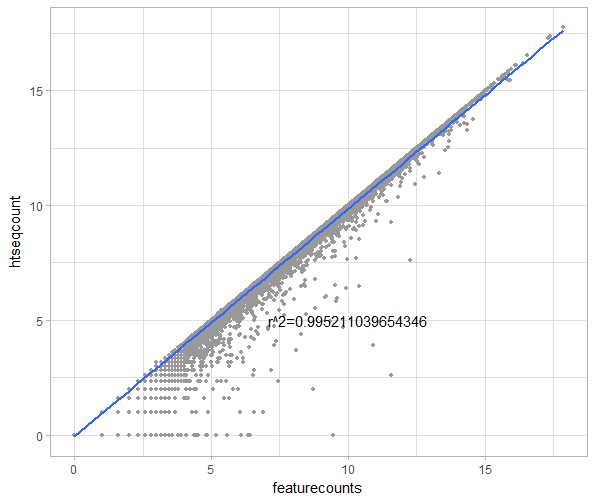
最后用R作个散点图看看两者同个基因的reads数目大小的分布(取log2),并计算个相关系数
library(ggplot2)
data <- read.table(file = "count.txt", sep = "\t", header = T, row.names = 1, stringsAsFactors = F)
data_matrix <- log2(data + 1)
model <- lm(featurecounts ~ htseqcount, data_matrix)
r2 <- summary(model)$r.squared
r2 <- paste0("r^2=", r2)
p <- ggplot(data_matrix, aes(x = featurecounts, y = htseqcount)) +
geom_point(colour = "grey60", size = 1) +
theme_light() +
stat_smooth(method = lm, se = FALSE) +
annotate("text", label = r2, x = 3.5, y = 1)
p从下图中可看出,两者的数据是呈正相关,两者绝大部分的counts数是非常接近的,在低表达量的那部分数据中HTSeq-count的值偏小于featureCounts。因而至少这个数据来说,我觉得featureCounts代替HTSeq-count进行表达量定量是完全没问题的。
本文已在《生信技能树》公众号上投稿,并授权转载
本文出自于http://www.bioinfo-scrounger.com转载请注明出处