rMATS是一款对RNA-Seq数据进行差异可变剪切分析的软件。其通过rMATS统计模型对不同样本(有生物学重复的)进行可变剪切事件的表达定量,然后以likelihood-ratio test计算P value来表示两组样品在IncLevel(Inclusion Level)水平上的差异(从公式上来看,IncLevel跟PSI的定义也是类似的),lncLevel并利用Benjamini Hochberg算法对p value进行校正得FDR值。rMATS可识别的可变剪切事件有5种,分别是skipped exon (SE)外显子跳跃,alternative 5' splice site (A5SS)第一个外显子可变剪切,alternative 3' splice site (A3SS)最后一个外显子可变剪切,mutually exclusive exons (MXE)外显子选择性跳跃和 retained intron (RI)内含子滞留,展现形式如下图(来自官网http://rnaseq-mats.sourceforge.net/index.html)
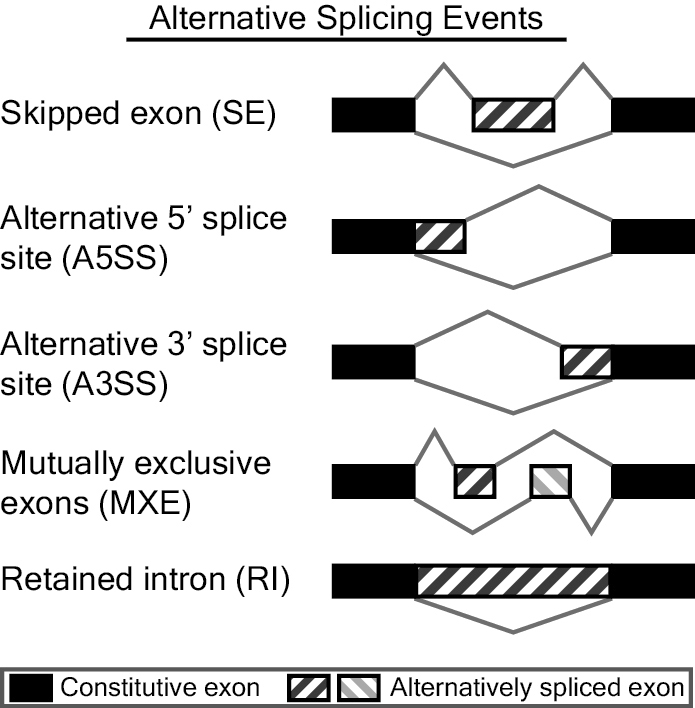
rMATS最近刚现在出了rMATS 4.0.1版,相比之间的rMATS 3.2.5版,其用C,Python,Cython重写了该软件,运算速度提升了100倍,并且可支持多线程执行(明显感觉到计算速度的提升),并且新版的安装也简便好多了。PS.老版的rMATS我那时都是用bioconda安装的,不然太折腾了。。
进官网或者下述网站进行下载
https://sourceforge.net/projects/rnaseq-mats/files/MATS/rMATS.4.0.1.tgz/download然后按照官网说明安装一些库文件以及python库(以ubuntu为例)
pip install numpy
sudo apt-get install libblas-dev liblapack-dev
sudo apt-get install gfortran如果python的numpy装的有问题,可以使用bioconda来装下旧版的rMATS,其会顺便把numpy也装好,然后将其放置在环境变量中就行了(一般也不用这样)
如果运行时报错:error while loading shared libraries: libgsl.so.0: cannot open shared object file: No such file or directory,则说明缺少libgsl.so.0库文件,按照下述安装下就好
sudo apt-get install libgsl0-dev软件的使用
软件使用也很简单,rMATS支持两种格式文件的输入。第一种是fastq格式,那么在安装的时候还需要安装STAR比对软件以及提供比对的索引文件(STAR的索引文件异常的大),所以rMATS其实是建议使用第二种方式;第二种是bam格式,rMATS支持其他比对软件比对后的结果bam文件作为输入,比如tophat等(那么hisat2也没啥问题,我试过),这样也能减少rMATS的运行时间。
新版rMATS下载解压后,你会发现有两个rmats.py执行脚本,这是由于rMATS v4.0.1 (turbo) was built with two different settings of Python interpreter,所以我们需要先测试下自己的系统支持那种,进入python,输入下述命令
>>> import sys
>>> print sys.maxunicode如果出现1114111则说明需要使用rMATS-turbo-Linux-UCS4文件夹下rmats.py;如果出现65535则说明使用rMATS-turbo-Linux-UCS2文件夹下rmats.py
rMATS的参数设置不多,我一般使用以下几个,其他具体可参考官网
--b1 b1.txt 输入sample1的txt格式的文件,文件内以逗号分隔重复样本的bam文件名
--b2 b2.txt 输入sample2的txt格式的文件,文件内以逗号分隔重复样本的bam文件名
-t readType 双端测序则readType为paired,单端测序则为single
--readLength
测序reads的长度 --gtf gtfFile 需要输入的gtf文件
--od outDir 所有输出文件的路径(文件夹)
--nthread
设置线程数 --cstat
The cutoff splicing difference. The cutoff used in the null hypothesis test for differential splicing(这个我一直不太理解是怎么卡的阈值,是在算法识别一些新的可变剪切的时候的差异性吗)
python rMATS-turbo-Linux-UCS4/rmats.py --b1 b1.txt --b2 b2.txt --gtf Homo_sapiens.Ensembl.GRCh37.72.gtf --od AS -t paired --readLength 151 --cstat 0.0001 --nthread 10结果文件
rMATS的结果文件是以各个可变剪切事件的分布的,主要由AS_Event.MATS.JC.txt,AS_Event.MATS.JCEC.txt,fromGTF.AS_Event.txt,JC.raw.input.AS_Event.txt,JCEC.raw.input.AS_Event.txt这几类;其中JC和JCEC的区别在于前者考虑跨越剪切位点的reads,而后者不仅考虑前者的reads还考虑到只比对到第一张图中条纹的区域(也就是说没有跨越剪切位点的reads),但是我们一般使用JC的结果就够了(如果只是单纯的比较两组样品间可变剪切的差异的话)。
这几类文件中比较重要的要数S_Event.MATS.JC.txt,因为其他文件的信息差不多最终都整合在这个文件里面,以SE.MATS.JC.txt为例:
1-5列看列名就能懂其意思的,分别ID,GeneID,geneSymbol,chr,strand
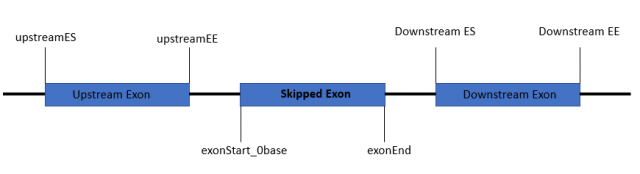
6-11列分别为外显子的位置信息,分别为exonStart_0base,exonEnd,upstreamES,upstreamEE,downstreamES,downstreamEE;网上有张图能很好的解释其含义,如下所示;其他可变剪切文件的这几列有点略微不同,但都可以类似的理解
12列又为ID,不知道为啥重复再来一次,可能为了布局美观吧。。。
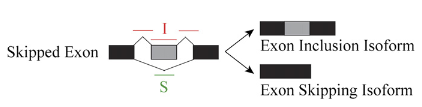
13-16列展示两组样品在inclusion junction(IJC)和skipping junction counts(SJC)下的count数,重复样本的结果以逗号分隔;列名分别为IJC_SAMPLE_1,SJC_SAMPLE_1,IJC_SAMPLE_2,SJC_SAMPLE_2,可以从下图来理解下
下面几列信息rMATS认为是极为重要的信息:
- lncFormLen :length of inclusion form, used for normalization
- SkipFormLen : length of skipping form, used for normalization
- P-Value : Significance of splicing difference between two sample groups(两组样品可变剪切的统计学显著差异指标)
- FDR : False Discovery Rate calculated from p-value(对p-value的FDR校正)
- lncLevel1 : inclusion level for SAMPLE_1 replicates (comma separated) calculated from normalized counts
- IncLevel2 : inclusion level for SAMPLE_2 replicates (comma separated) calculated from normalized counts
- IncLevelDifference : average(IncLevel1) - average(IncLevel2)
lncFormLen和SkipFormLen分别是inclusion form和skipping form的有效长度值,虽然有计算公式但是还是要根据reads跨越时的具体情况来定的,具体解释可见https://groups.google.com/forum/#!topic/rmats-user-group/d7rzUBKXF1U(需翻墙。。。才能看哦)
lncLevel1和IncLevel2分别为sample1和sample2的inclusion level(粗略可理解为PSI),从公式上看比较好理解
ψ = (I/LI) / (I/LI + S/LS)ψ = Inclusion Level(sample1),I = IJC_SAMPLE_1,S = SJC_SAMPLE_1,LI = lncFormLen,LS = SkipFormLen
从公式上理解为lncLevel是在最终的成熟mRNA中,这个respective exon(SE事件中则是对应那个被跳跃的exon)出现的平均频率是多少,或者说所占的比例?也可以认为是read counts(标准化后的)在可变剪切的事件中的各自exon上所占的比例?简单的说就是lncLevel越小说明出现外显子的跳跃的比例越高(这个是以SE文件为例的)???可能我还理解的不是很准确。。。。
最后还有一个软件能绘制可变剪切的图片的软件rmats2sashimiplot
参考资料:
https://www.biostars.org/p/256949/#274012
http://rnaseq-mats.sourceforge.net/user_guide.htm#as_events
本文已在《生信技能树》公众号上投稿,并授权转载
本文出自于http://www.bioinfo-scrounger.com转载请注明出处